
| 背景
本應用文獻概述了有關生物製劑許可申請 (BLA,Biologics License Applications) 的可用指南,以及由於內毒素測試方法修改或鱟試劑 (LAL,Limulus Amoebocyte Lysate) 供應商變化,如何實行變更。生物製劑許可申請針對藥品,因此適用於成品藥測試,而非製程中的樣品或水樣。本文還探討了質量控制 (QC) 部門內的工作流程變更。
完成了內毒素測試驗證的成品藥只有在向監管部門申報之後方可出廠,因而必須向監管部門申報測試方法或鱟試劑供應商的詳細變更內容。請注意,在重新驗證藥品時,在申報中標註通用試劑或方法的公司,比標註特定試劑或方法的公司 (例如:標註「FDA 許可供應商」或「藥典光度法」) 通常會有更大的靈活性,有更多的可用方法和試劑選項。
在進行變更時,還須考慮公司內部的需求。大多數質量控制實驗室和質量保證部門在進行變更時都會遵守特定的標準操作規程 (SOP) 和公司的質量管理體系 (QMS)。在變更之前,通常先建立變更控制 (Change Control) 程序,並由跨職能部門出具詳細的變更和評估文檔。一旦決定變更,就啟動變更程序,質量控制實驗室啟動重新驗證程序以更改內毒素測試方法或鱟試劑。
美國藥典 (USP) 第 <85> 章和歐洲藥典 (EP) 第 2.6.14 節規定,「當發生任何可能影響測試結果的條件變化時,都必須重新測試干擾因素」[1][2]。
在更改測試方法或鱟試劑供應商時,必須進行干擾因素測試或「產品篩選/驗證」[1]。通常以不同的稀釋度來篩選產品,以確定適用於新的測試方法或鱟試劑供應商的最佳稀釋度。一旦確定了最佳稀釋度,應測試三個離散批次的產品,以在新的條件下完成驗證。如果實驗室要從 96 孔板顯色測試過渡到同樣使用顯色法的其它平台,由於測試的生物化學特性沒有變化,建議對先前驗證的稀釋度進行單批次驗證。
| 法規和指南
監管部門和行業指導文件都未對重新驗證產品給出明確建議。本文將找出可用的建議,並指出建議的出處。
鱟試劑有不同的配方,配方因供應商而異。當公司打算更換鱟試劑供應商,並想知道更換供應商後是否需要重新驗證產品時,卻從通用的 USP、EP、JP 章節中找不到明確答案。由美國國家標準學會 (ANSI,American National Standards Institute) 認證的醫療儀器促進協會 (AAMI,Association for the Advancement of Medical Instrumentation) 在其文檔的第 ST72:2019 章節中提供了一些有關更改鱟試劑供應商的指南。該章明確指出,如果更改細菌內毒素測試 (BET,Bacterial Endotoxins Testing) 試劑的來源,或更改細菌內毒素測試技術 (例如:從凝膠法改為動態顯色法),就須重新進行評估或適用性研究 [3]。此章雖然適用於醫療器械,但 FDA 表示,如果公司決定進行上述更改,可以遵照此章的指南。
如果打算更改鱟試劑供應商或內毒素測試方法,必須申報該變更,或將變更包含在年度報告中。應採用哪種方式,取決於變更類型 (例如:變更鱟試劑供應商或變更測試方法等)。鱟試劑測試是藥品出廠的關鍵性測試,因此申請生物製劑許可的公司必須在文檔材料中包含鱟試劑測試。在重新驗證產品時,由於需要採用不同的測試方法或內毒素試劑,因而申報工作可能很麻煩。 FDA 在行業指導文件「已批准的新藥或簡略新藥的變更 (Changes to An Approved NDA or ANDA)」的「規範 (Specifications)」一章提供了有關申報變更的信息。該文件說明了以下兩種與鱟試劑測試相關的變更申報:較小變更的申報 (Minor Filing Change) 和中度變更的申報 (Moderate Filing Change)。有關內毒素測試方法或鱟試劑供應商的變更申報示例,請參見 圖 1。
較小變更的申報
可以在提交給 FDA 的年度報告中說明較小變更的內容。較小變更 (例如:在保持動態顯色法的情況下變更鱟試劑供應商) 對藥品的影響不大。對於較小變更,公司只需提交「可比擬任務 (Comparability Protocol)」來說明測試、研究、結果,以顯示新的鱟試劑供應商的合格性。新藥、簡略新藥、生物製劑的許可申請都需要提交年度報告,因此公司無需花時間來另行申報較小變更。
中度變更的申報
中度變更的申報有以下兩種:
- 變更生效期 (CBE,Changes Being Effective) 在 30 天內:要求公司在分銷涉及變更的藥品之前的 30 天內向 FDA 提交補充材料。此補充材料應明確標註「補充材料 – 變更生效期在 30 天內 (Supplement – Changes Being Effective in 30 Days)」。如果 FDA 在收到補充材料後 30 天內告知申請人缺失部分信息,則申請人必須推遲分銷藥品,直到在補充材料中提供缺失的信息。
- 變更生效期在 0 天內 (即立即生效):變更生效期在 0 天內的補充材料包括某些中度變更,允許公司在 FDA 收到補充材料後立即分銷藥品。如果變更了鱟試劑供應商或測試方法,FDA 會在新方法符合 USP <85> 要求的情況下批准鱟試劑供應商的變更申請。
從一種鱟試劑測試方法更改為另一種鱟試劑測試方法 (例如:從凝膠法改為動態顯色法) 通常被作為生效期在 0 天內 (CBE-0) 的變更來提交申報。對於此類變更,公司只需提交「可比擬任務」來說明測試、研究、結果,以顯示新的測試方法的有效性。而生效期在 30 天內 (CBE-30) 的變更申報則較為保守,因為這會給 FDA 充足的時間來審查變更。
| 總結
本應用文獻概述了關於細菌內毒素測試的 FDA 文件和藥典章節中的建議和指南。但請注意,這些文件都未給出明確的變更申報方式。質量控制實驗室應諮詢本公司的規則監管部門和質量保證部門,並根據公司的質量管理體係來確定最適合的變更。圖1 是關於內毒素測試方法的變更示例,以及最適合的申報方式。可以在公司的年度報告中包括變更申報,也可以直接向 FDA 提交變更通知。
重新驗證產品以確認內毒素測試系統是創新性的和完全符合監管要求的,這項工作並非想像中的那樣麻煩。 Sievers* Eclipse 細菌內毒素測試平台具有簡化工藝流程的任務功能,大大提高了質量控制實驗室在變更測試平台時的工作效率。變更管理的前期投入,很快就會在自動化的測試工作中得到回報。 Eclipse 提高了實驗室的工作效率,減少了培訓工作量,簡化了驗證工作,從而大大降低了生產成本、節省了時間。
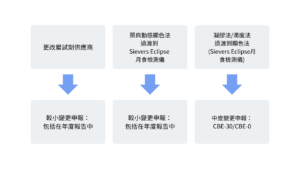
圖 1:生物製劑許可申請變更示例
參考文獻:
[1]. USP <85> Bacterial Endotoxins Test
[2]. EP 2.6.14 Bacterial Endotoxins
[3]. ANSI/AAMI ST72: Bacterial endotoxins – Test Methods, routine, monitoring, and alternatives to batch testing。章節/步驟 9.6.1.2,第 10 頁



